
# Importando a biblioteca BioPython
from Bio import SeqIO
import sys
# Definindo a entrada e a saída dos arquivos
entrada = sys.argv[1]
saida = sys.argv[2]
# Realizando a conversão dos dados
records = SeqIO.parse(entrada, "abi")
count = SeqIO.write(records, saida, "fastq")Processando dados de sequenciamento Sanger
Bioinformatics
Tutorial
Sanger
Resumo
Atividade Prática realizada no dia 05/11/2025, na disciplina de “Filogenia Molecular”, ministrada pelo Prof. Marco A. Costa, na Universidade Estadual de Santa Cruz (UESC) pelo Programa de Pós-Graduação em Genética e Biologia Molecular.
Palavras-chave
Árvore Filogenética, Evolução, Métdos Moleculares
1 Atividade
Editar as sequências contidas nos eletroferogramas anexos, gerando um arquivo word com as sequências Forward e Reverse, e as sequências consenso. Verificar qual espécie no banco de dados do NCBI apresenta maior identidade com cada sequência consenso obtida.
2 Metodologia
A atividade deveria ser realizada a partir do software BioEdit, entretanto não foi possível encontrar binários do software que fossem suportados pelo sistema operacional Linux (Archinux), que atualmente está sendo utilizado pelo discente.
Para contornar a situação, serão adotadas estratégias de processamento para processar os dados resultantes do sequenciamento Sanger (eletroferograma), que sigam os mesmos passos do tutorial apresentado em sala de aula.
2.1 Análise dos arquivos .ab1
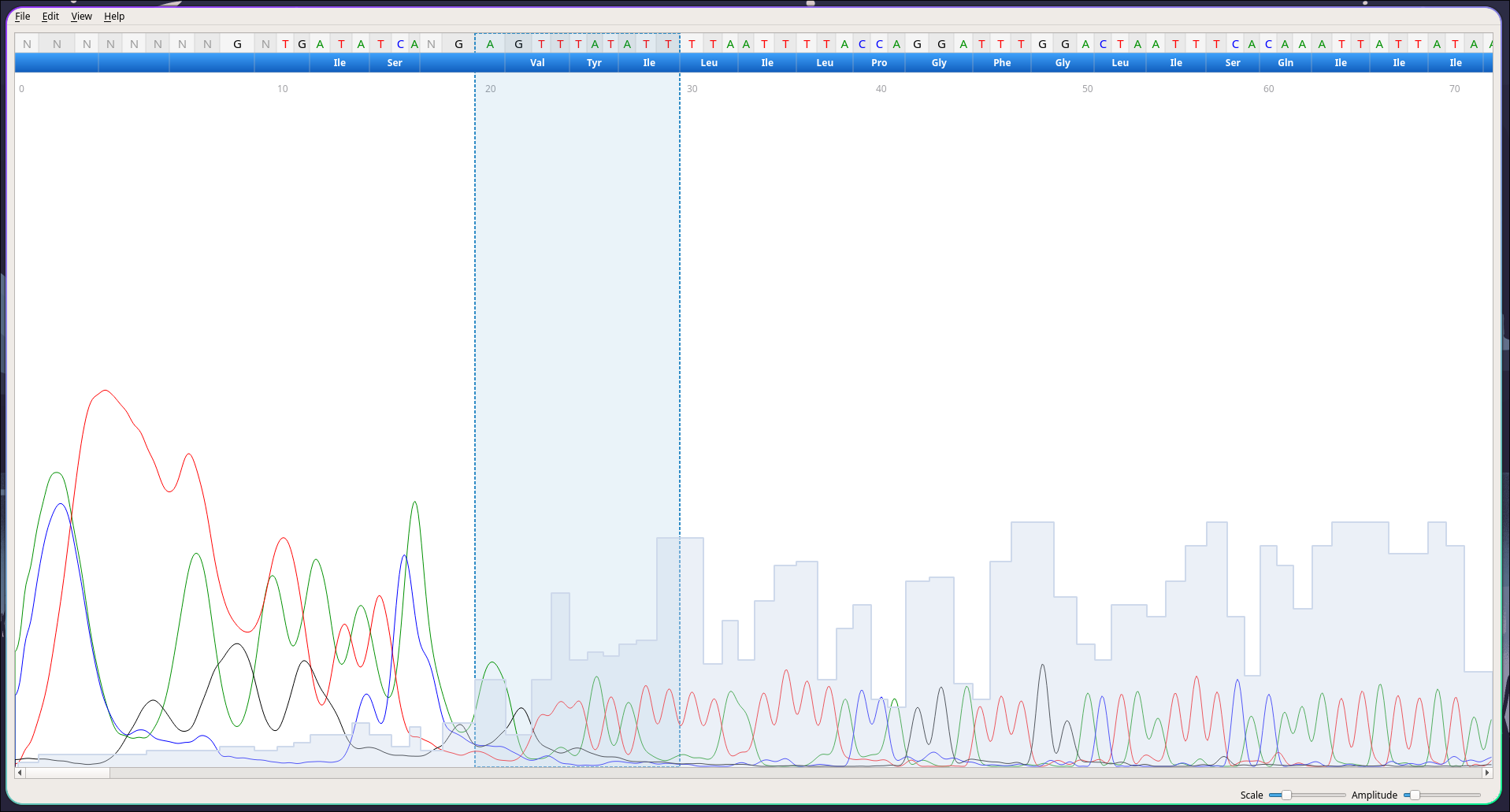
Arquivos .ab1 são dados brutos vindos de sequenciadores tipo Sanger. Esses arquivos possuem a sequência do DNA analisada, um eletroferograma (um gráfico mostrando os picos de fluorenscência para cada base) e uma pontuação de qualidade associada à cada base da sequência.
Nesse caso vamos utilizar o software CutePeaks para realizar o basecalling, a chamada das bases individuais da sequência analisada, para conferir seu pico de fluorescência e sua qualidade.
2.2 Conversão de .ab1 para .fastq
Como não será possível trabalhar diretamente com o software BioEdit, será necessário realizar a conversão dos arquivos arquivos .ab1 para o formato .fastq, formato padrão dos dados brutos de sequenciadores de nova gereação, como os da plataforma Illumina.
Para realizar essa conversão, foi utilizada a biblioteca BioPython (suportada pela linguagem de programação Python) para criar o arquivo abi2fastq.py com o seguinte código:
O arquivo abi2fastq.py pode ser utilizado para inserir os dois argumentos para a conversão, o nome do aruivo de entrada e o nome do arquivo de saída (que será a conversão). Para rodar o código e realizar a conversão, executa-se no terminal linux:
python abi2fastq.py COC_2F.ab1 COC_2F.fastq
Com isso teremos nosso arquivo .ab1 convertido para formato .fastq, seguindo essa estrutura:
@13F
NNNNNNNNGNTGATATCANGAGTTTATATTTTAATTTTACCAGGATTTGGACTAATTTCACAAA
TTATTATAAATGAAAGAGGAAAAAAGGAAATTTTTGGAAATTTAAGAATAATTTATGCTATATT
AGGAATTGGATTTTTAGGATTTATTGTATGAGCTCATCATATATTTACTGTAGGATTAGATGTT
GATACACGAGCATATTTTACATCTGCAACAATAATTATTGCAATTCCTACAGGAATTAAAGTTT
TTAGATGATTAGCAACTTATCATGGATCAAAATTAAATTTTAATATTTCATTTATATGATCAAT
TGGATTTATTTTAATATTTACTATTGGAGGATTAACAGGAATTATATTATCAAATTCATCAATT
GATATTATTTTACATGATTCTTATTACGTAGTTGGTCATTTTCACTATGTATTATCTATAGGAG
CAGTATTTTCCATTATTGCAAGATTTATTCATTGATTTCCCTTATTATCAGGATTAATAATTAA
TCAAAAATGATTAAAATTTCAATTTTTTTTTATATTCATTGGAATTAATTTCACTTTTTTTCCT
CAACATTTTTTAGGATTAATATCAGATCAAATGGAAATTACGGCACTTATGGATAATACATAGT
GAAAATGACCAACTCCGTATTAAGACTCATTGTATCAT
+
"$$$$%%%&%&')),)&+%,7)7M<>=@A[[;F<KTU7DJ20PQ62U__L@<JJGPY_G8YTIY
___WW_Y99H8G=<EL_\_\Y\\O\____\\_\\\_Y\\\\__\\\_____\___\_\\\\___
\HYWYR__\RG==LL\\_\\__W___\\___TYY\RYQAY\\Y___W___T\T_\\T\=IOYEQ
WOYY\LTDLCELTSDQQ@YLEYY_____________________W________\__________
___\___\\________________W_____________________W_________W______
___\__________________________\\_____________________RR_________
_\______________\_______________________________________________
______________________W_______YFF_W_O__O____________PW__________
_______W__W_________________OO______WPOO_L_[[[[[[[[[U[[[[[[[[[[W
[QUWUWW[[[LLUW[UIWKPUJG-&&,5H''*130/,3B:,8K/**'.(9(>3@/',-*'+*?(
4+*2/-J****'*(+(43-*3/*<''/**1(B+2*+)(Onde o identificador da sequência é o @13F e a pontuação Phred de qualidade está localizada abaixo do símbolo de +. Em termos práticos esse arquivo informa a sequência e a qualidade das bases, mas diferente do formato anterior, os picos de fluorescência não são informados.
2.3 Checagem da qualidade dos arquivos fastq
Para checar se a conversão foi realizada corretamente, os arquivos de qualidade das sequências .fastq foram comparadas com a qualidade exibida no eletroferograma e checada no CutePeaks. Essa checagem foi realizada com o software FastQC.
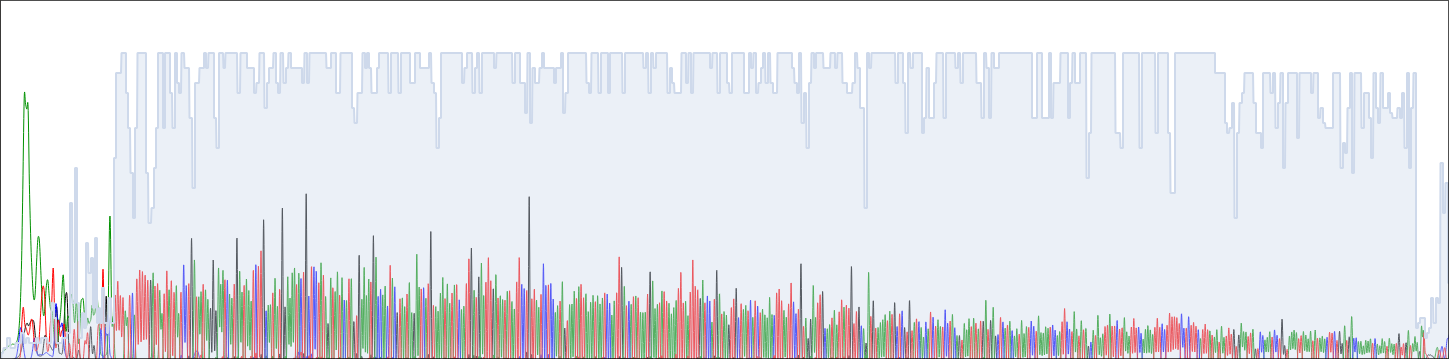
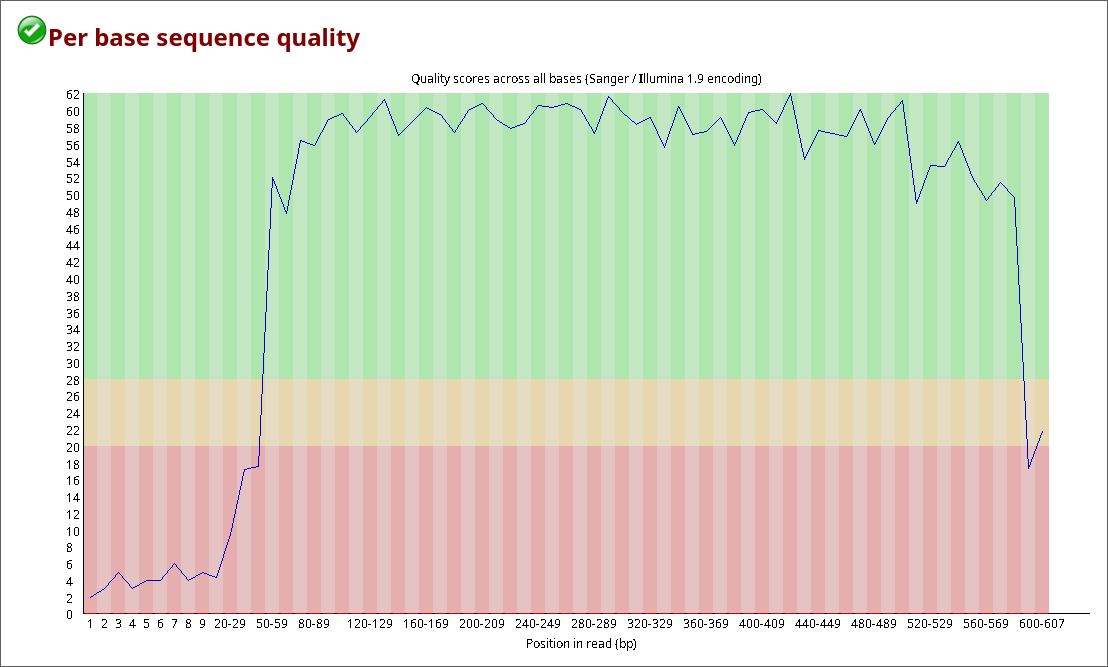
2.4 Remoção das bases de baixa qualidade
Para a remoção das bases de baixa qualidade foi utilizado o software Seqtk, em sua função trimfq, que remove automaticamente as sequências de baixa qualidade (Phred < 15) e bases não definidas.
As análises de quailidade também foram realizadas nas sequências após a remoção das bases de baixa qualidade.
2.5 Conversão de FASTQ para FASTA
Após a trimagem das sequências e remoção de bases de não definidas, os arquivos FASTQ foram comvertidos para o formato FASTA através da biblioteca BioPython, seguindo a mesma lógica da conversão dos arquivos .ab1:
# Importando a biblioteca BioPython
from Bio import SeqIO
import sys
# Definindo a entrada e a saída dos arquivos
entrada = sys.argv[1]
saida = sys.argv[2]
# Realizando a conversão dos dados
records = SeqIO.parse(entrada, "fastq")
count = SeqIO.write(records, saida, "fasta")O arquivo final fastq2fasta.py então é chamado no terminal linux:
python fastq2fasta.py COC_2F.fastq COC_2F.fasta
Um arquivo em formato FASTA se assemelha à um arquivo FASTQ, porém sem os índices de qualidade por base (Phred Score) anexados ao arquivo das sequências. Outra diferença são os símbolos adotados, o header de uma sequência (seu cabeçalho) agora é marcado pelo símbolo > antes do nome da sequência.
2.6 Obtenção da Sequência Consenso
A sequência conseso é o resultado da união via sobreposição das sequências foward e reverse do sequenciemanto. Para fazer a montegem dessas sequências em uma única final. Para isso pode-se utilizar o software CAP3, que alinha a sobreposição das sequências para estabelecer as contigs finais, uma técnica denominada de Overlap Layout Consensus.
A entrada para o software CAP3 foi um arquivo FASTA com a junção das duas sequências (foward e reverse) de cada amostra, dessa maneira (Arquivo COC_merge.fasta):
>13F
TTTATATTTTAATTTTACCAGGATTTGGACTAATTTCACAAATTATTATAAATGAAAGAG
GAAAAAAGGAAATTTTTGGAAATTTAAGAATAATTTATGCTATATTAGGAATTGGATTTT
TAGGATTTATTGTATGAGCTCATCATATATTTACTGTAGGATTAGATGTTGATACACGAG
CATATTTTACATCTGCAACAATAATTATTGCAATTCCTACAGGAATTAAAGTTTTTAGAT
GATTAGCAACTTATCATGGATCAAAATTAAATTTTAATATTTCATTTATATGATCAATTG
GATTTATTTTAATATTTACTATTGGAGGATTAACAGGAATTATATTATCAAATTCATCAA
TTGATATTATTTTACATGATTCTTATTACGTAGTTGGTCATTTTCACTATGTATTATCTA
TAGGAGCAGTATTTTCCATTATTGCAAGATTTATTCATTGATTTCCCTTATTATCAGGAT
TAATAATTAATCAAAAATGATTAAAATTTCAATTTTTTTTTATATTCATTGGAATTAATT
TCACTTTTTTTCCTCAACATTTTTTAGGATTAATATC
>13R
AAAAAAAAATTGAAATTTTAATCATTTTTGATTAATTATTAATCCTGATAATAAGGGAAA
TCAATGAATAAATCTTGCAATAATGGAAAATACTGCTCCTATAGATAATACATAGTGAAA
ATGACCAACTACGTAATAAGAATCATGTAAAATAATATCAATTGATGAATTTGATAATAT
AATTCCTGTTAATCCTCCAATAGTAAATATTAAAATAAATCCAATTGATCATATAAATGA
AATATTAAAATTTAATTTTGATCCATGATAAGTTGCTAATCATCTAAAAACTTTAATTCC
TGTAGGAATTGCAATAATTATTGTTGCAGATGTAAAATATGCTCGTGTATCAACATCTAA
TCCTACAGTAAATATATGATGAGCTCATACAATAAATCCTAAAAATCCAATTCCTAATAT
AGCATAAATTATTCTTAAATTTCCAAAAATTTCCTTTTTTCCTCTTTCATTTATAATAAT
TTGTGAAATTAGTCCAAATCCTGGTAAAATTAAAATATAAACTTCTGGATGACCAAAAAA
TCAAAATAAATGTTGATAA2.7 Alinhamento das sequnências
As sequências finais foram Alinhadas via BLASTn online contra o banco de dados core_nt, uma versão reduzida e clusterizada do banco total de nucleotídeos nt.
3 Resultados
3.1 Qualidade das Sequências
A qualidade sequências dos arquvos FASTQ foram realizadas com o software FastQC e depois agragadas com o software MultiQC. As qualidades foram medidas antes e depois da remoção de sequências de baixa qualidade.
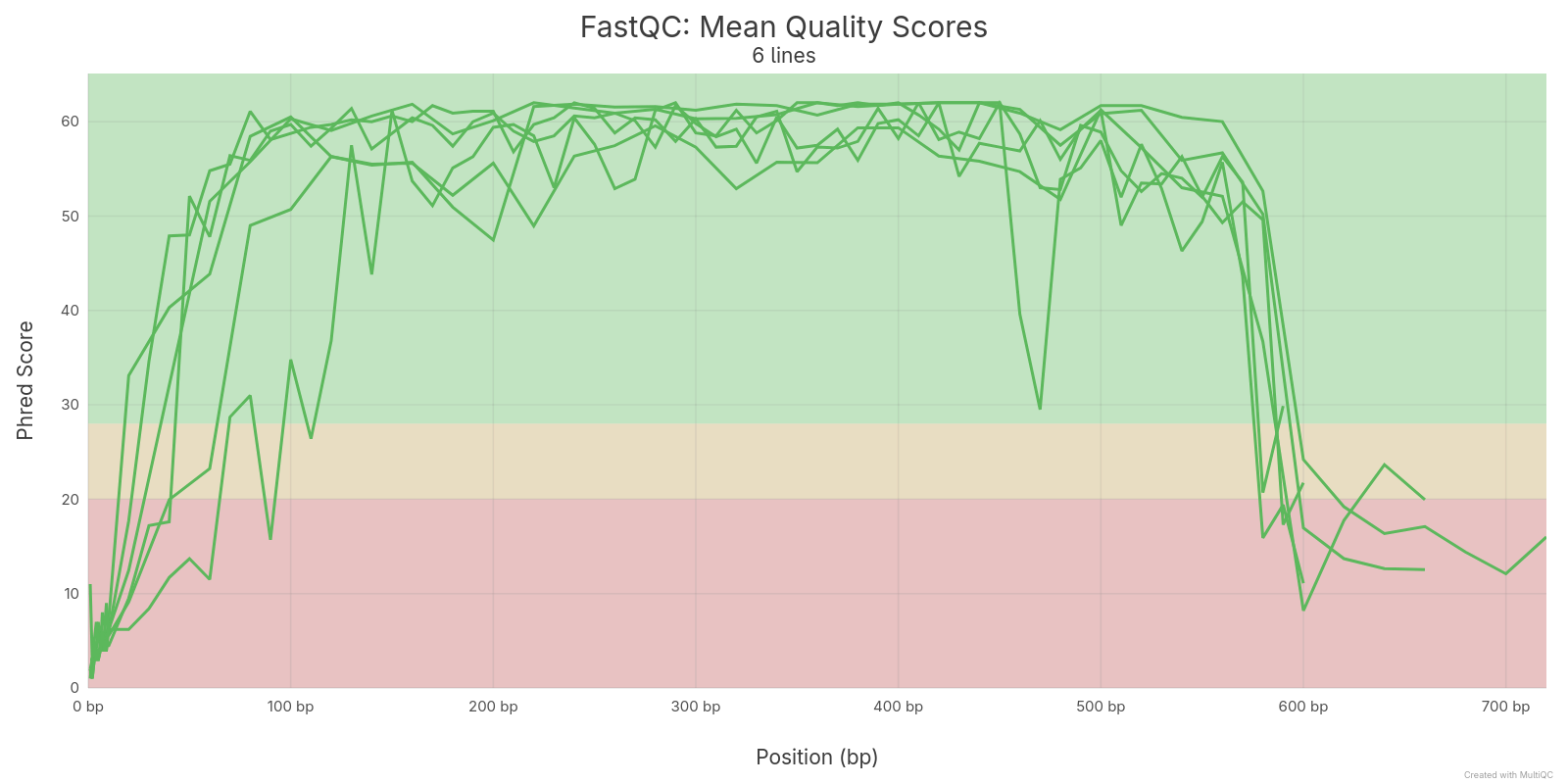
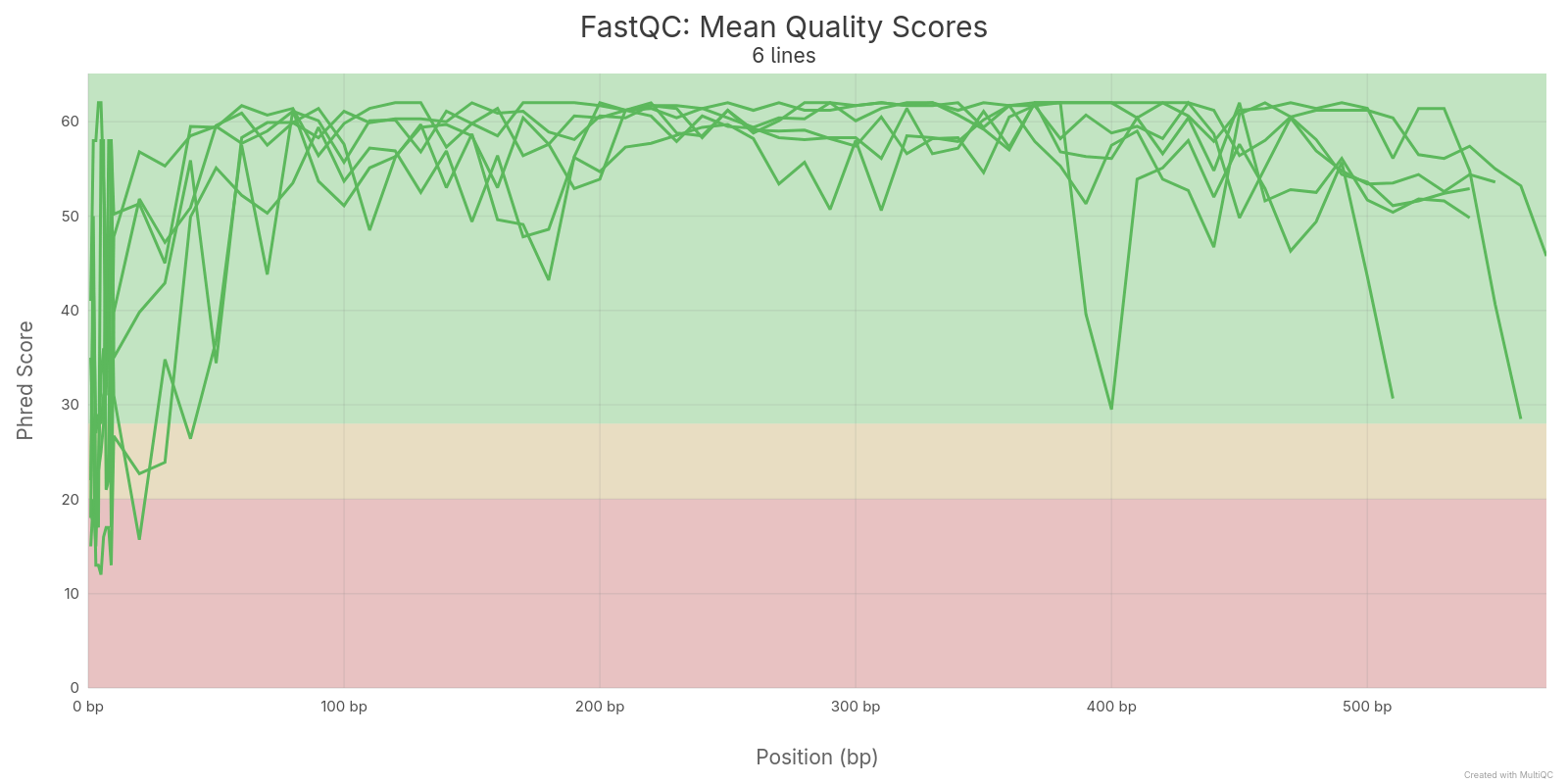
3.2 Sequências montadas
As sequências fasta (foward e reverse) foram unidas em arquivos únicos para cada código e amostra, COC_merged.fasta; MAC_merged.fasta e RBA_merged.fasta. Esses arquivos foram montados utilizando o software CAP3 e tiveram como resultado final os seguintes tamanhos em bases:
| Amostra | Tamanho (nt) |
|---|---|
| COC | 615 |
| MAC | 607 |
| RBA | 617 |
Como resultado da montagem, temos o arquivo FASTA final para cada sequência:
>COC
TTATCAACATTTATTTTGATTTTTTGGTCATCCAGAAGTTTATATTTTAATTTTACCAGG
ATTTGGACTAATTTCACAAATTATTATAAATGAAAGAGGAAAAAAGGAAATTTTTGGAAA
TTTAAGAATAATTTATGCTATATTAGGAATTGGATTTTTAGGATTTATTGTATGAGCTCA
TCATATATTTACTGTAGGATTAGATGTTGATACACGAGCATATTTTACATCTGCAACAAT
AATTATTGCAATTCCTACAGGAATTAAAGTTTTTAGATGATTAGCAACTTATCATGGATC
AAAATTAAATTTTAATATTTCATTTATATGATCAATTGGATTTATTTTAATATTTACTAT
TGGAGGATTAACAGGAATTATATTATCAAATTCATCAATTGATATTATTTTACATGATTC
TTATTACGTAGTTGGTCATTTTCACTATGTATTATCTATAGGAGCAGTATTTTCCATTAT
TGCAAGATTTATTCATTGATTTCCCTTATTATCAGGATTAATAATTAATCAAAAATGATT
AAAATTTCAATTTTTTTTTATATTCATTGGAATTAATTTCACTTTTTTTCCTCAACATTT
TTTAGGATTAATATC
>RBA
TTATCAACATTTATTTTGATTTTTTGGTCATCCAGAAGTTTATATTTTAATTTTACCAGG
ATTTGGACTAATTTCACAAATTATTATAAATGAAAGAGGAAAAAAGGAAATTTTTGGAAA
TTTAAGAATAATTTACGCTATATTAGGAATTGGATTTTTAGGATTTATTGTATGAGCTCA
TCATATATTTACTGTAGGATTAGATGTTGATACACGAGCATATTTTACATCTGCAACAAT
AATTATTGCAATTCCTACAGGAATTAAAGTTTTTAGATGATTAGCAACTTATCATGGATC
AAAATTAAATTTTAATATTTCATTTATATGATCAATTGGATTTATTTTAATATTTACTAT
TGGAGGATTAACAGGAATTATATTATCAAATTCATCAATTGATATTATTCTACATGATTC
TTATTACGTAGTTGGTCATTTTCACTATGTATTATCTATAGGAGCAGTATTTTCCATTAT
TGCAAGATTTATTCATTGATTTCCCTTATTATCAGTGATTAATAATTAATCAAAAATGAT
TAAAATTTCAATTTTTTTTTATATTCATTGGAATTAATTTAACTTTTTTTCCTCAACATT
TTTTAGG
>MAC
TTATCAACATTTATTTTGATTTTTTGGTCATCCAGAAGTTTATATTTTAATTTTACCAGG
ATTTGGACTAATTTCACAAATTATTATAAATGAAAGAGGAAAAAAGGAAATTTTTGGAAA
TTTAAGAATAATTTATGCTATATTAGGAATTGGATTTTTAGGATTTATTGTATGAGCTCA
TCATATATTTACTGTAGGATTAGATGTTGATACACGAGCATATTTTACATCTGCAACAAT
AATTATTGCAATTCCTACAGGAATTAAAGTTTTTAGATGATTAGCAACTTATCATGGATC
AAAATTAAATTTTAATATTTCATTTATATGATCAATTGGATTTATTTTAATATTTACTAT
TGGAGGATTAACAGGAATTATATTATCAAATTCATCAATTGATATTATTTTACATGATTC
TTATTACGTAGTTGGTCATTTTCACTATGTATTATCTATAGGAGCAGTATTTTCCATTAT
TGCAAGATTTATTCATTGATTTCCCTTATTATCAGGATTAATAATTAATCAAAAATGATT
AAAATTTCAATTTTTTTTTATATTCATTGGAATTAATTTCACTTTTTTTCCTCAACATTT
TTTAGGATTTAATATCA
3.3 Alinhamento no BLAST
Foi realizado o alinhamento global das sequências em um banco de dados de nucleotídeos (NCBI Core NT), os resultados foram os seguintes:
| Amostra | Melhor Hit | Organismo |
|---|---|---|
| COC | H2 cytochrome c oxidase subunit I (COI) gene | Partamona rustica |
| MAC | H2 cytochrome c oxidase subunit I (COI) gene | Partamona rustica |
| RBA | H5 cytochrome c oxidase subunit I (COI) gene | Partamona rustica |
Os alinhamentos tiveram índices de identidade e cobertura maiores que 99% para os melhores alinhamentos.
As sequências aparentam ser derivadas da espécie Partamona rustica, especificamente vindas de subunidades H do gene citocromo c oxidase, sendo uma enzima na cadeia respiratória de transporte de elétrons das células.